7.11 数据创建 EMP_mutate
模块EMP_mutate功能十分强大,不仅可以根据数据的assay,rowdata和coldata对数据列进行变构,还可以对数据分析结果进行变构。为了便于用户理解,以下对模块EMP_mutate的基本参数进行介绍:
- obj : 指定待分析的对象MAE或者EMPT。
- experiment : 指定待分析组学项目的名称(character)。
- ... :继承于dplyr包的mutate函数,可以支持各种数据变换。
- .by : 继承于dplyr包的mutate函数,相当于变换前执行了group_by()函数.
- .before : 继承于dplyr包的mutate函数, 设置新创建列的位置。
- .after : 继承于dplyr包的mutate函数, 设置新创建列的位置。
- mutate_by: 设置数据变换是针对sample还是feature。
- location:设置创建列是在assay, rowdata, 或者coldata。
- action: 设置“colwise”与“rowwise”选项,用于指示操作应沿列方向还是行方向执行。
- keep_result: 当输入值为 TRUE 时,系统将保留所有分析结果,样本与特征的任何更改均不影响此设置。当输入值为某个名称时,则仅该名称所对应的分析结果会被予以保留。
7.11.1 对全组学项目的表型数据进行创建变构
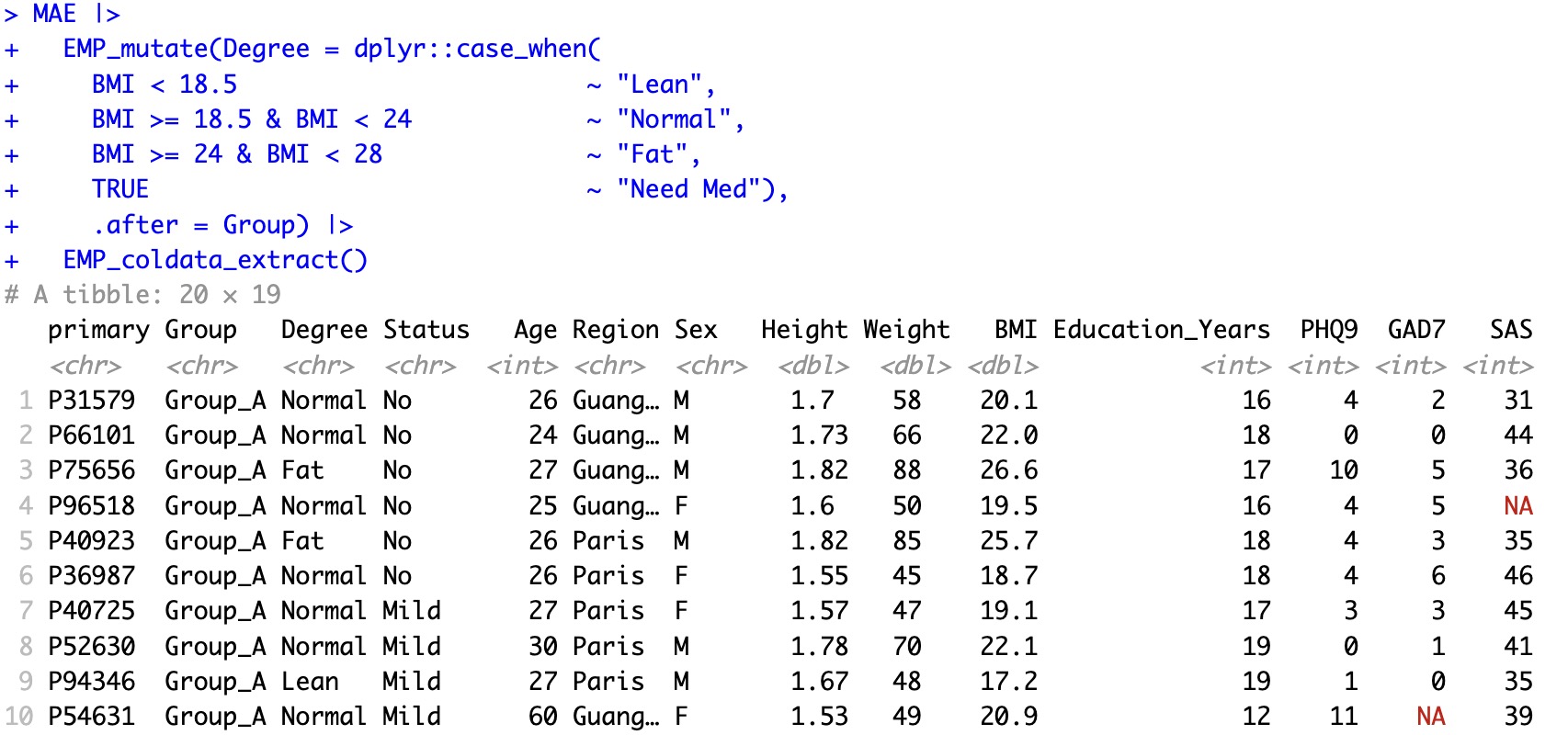
🏷️示例1: 根据BMI的数据,设置新的分组。
MAE |>
EMP_mutate(Degree = dplyr::case_when(
BMI < 18.5 ~ "Lean",
BMI >= 18.5 & BMI < 24 ~ "Normal",
BMI >= 24 & BMI < 28 ~ "Fat",
TRUE ~ "Need Med"),
.after = Group)
变换完成后,可以使用模块EMP_coldata_extract观察过滤后样本情况。
MAE |>
EMP_mutate(Degree = dplyr::case_when(
BMI < 18.5 ~ "Lean",
BMI >= 18.5 & BMI < 24 ~ "Normal",
BMI >= 24 & BMI < 28 ~ "Fat",
TRUE ~ "Need Med"),
.after = Group) |>
EMP_coldata_extract()
7.11.2 对单一组学项目数据及分析结果进行数据变换
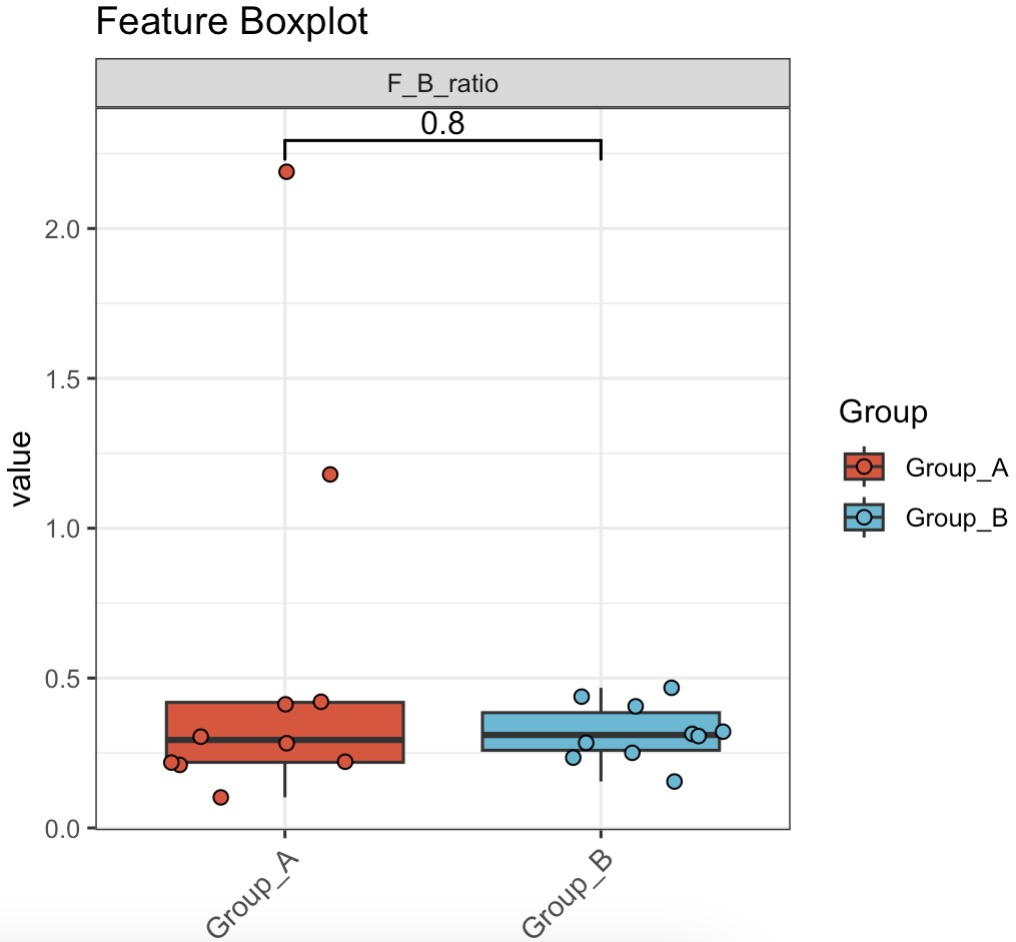
🏷️示例1: 在微生物组学数据中,创建Firmicutes/Bacteroidetes的比值特征。
MAE |>
EMP_assay_extract('taxonomy') |>
EMP_collapse(collapse_by = 'row',estimate_group = 'Phylum') |>
EMP_mutate(F_B_ratio = Firmicutes/Bacteroidetes,
.by = primary,.before = 2,
mutate_by = 'sample',location ='assay') |>
EMP_filter(filterFeature = 'F_B_ratio') |>
EMP_boxplot(estimate_group='Group')
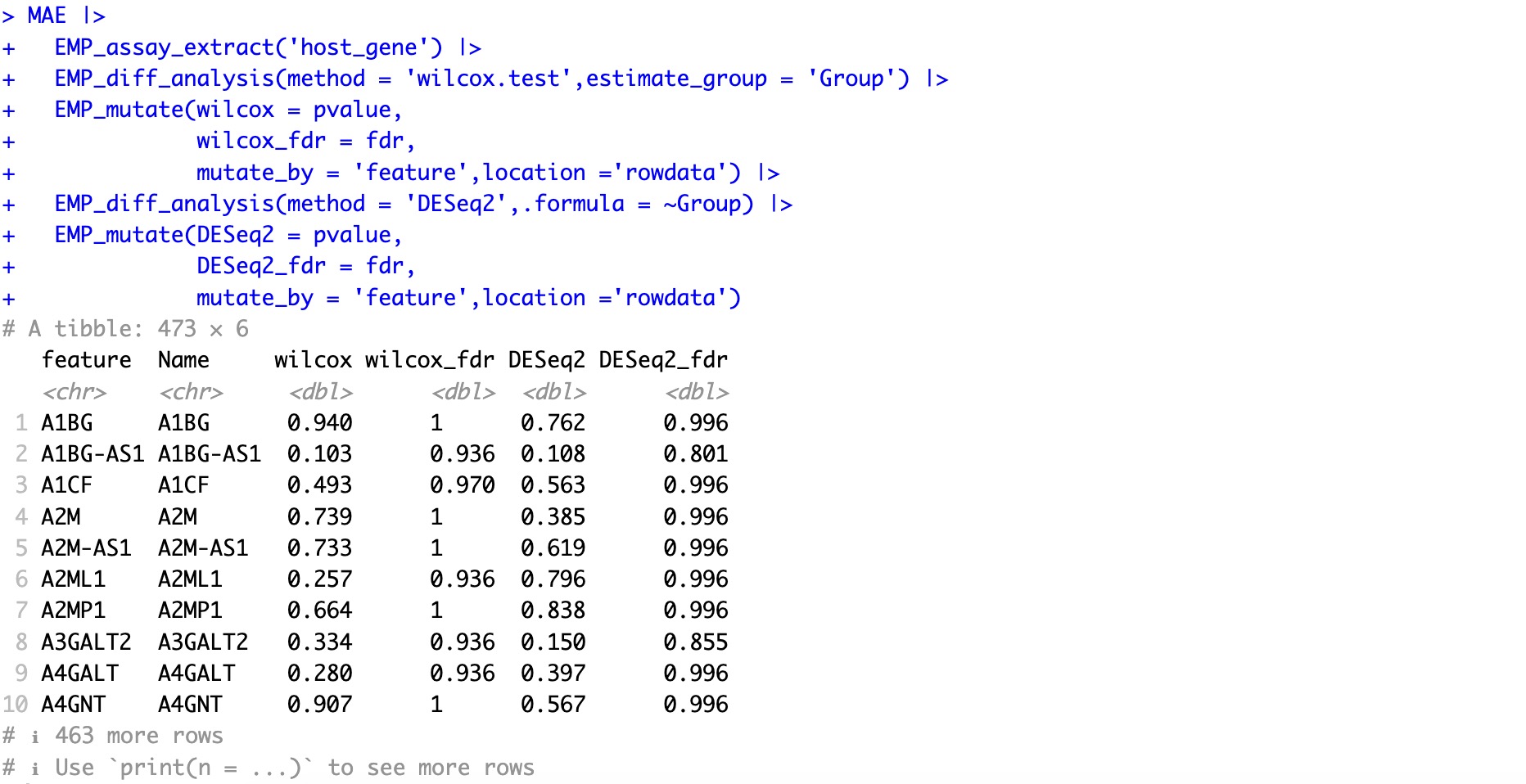
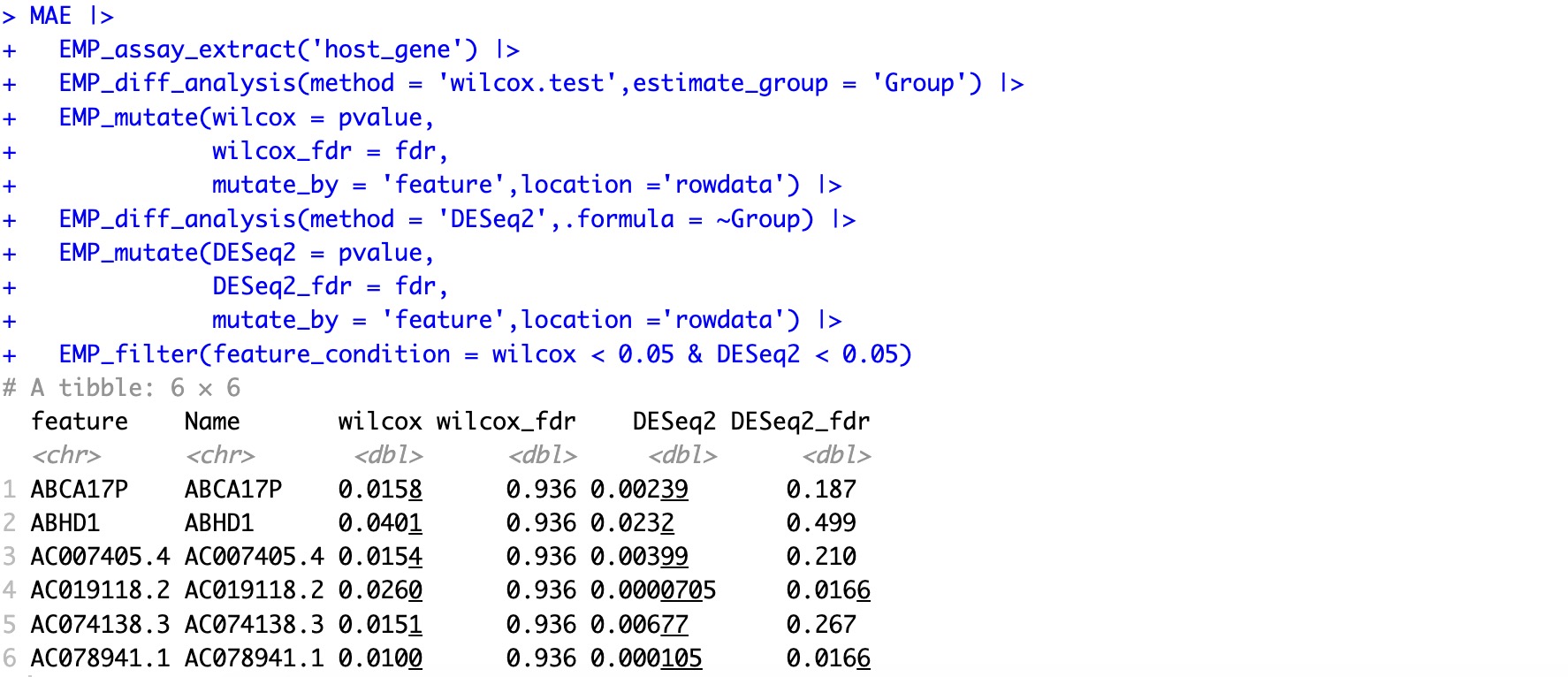
🏷️示例2:根据多种差异分析算法,合并其结果,并为特征增加注释信息。
MAE |>
EMP_assay_extract('host_gene') |>
EMP_diff_analysis(method = 'wilcox.test',estimate_group = 'Group') |>
EMP_mutate(wilcox = pvalue,
wilcox_fdr = fdr,
mutate_by = 'feature',location ='rowdata') |>
EMP_diff_analysis(method = 'DESeq2',.formula = ~Group) |>
EMP_mutate(DESeq2 = pvalue,
DESeq2_fdr = fdr,
mutate_by = 'feature',location ='rowdata')
这样我们就可以筛选出两种差异方法都显著的特征基因了。
MAE |>
EMP_assay_extract('host_gene') |>
EMP_diff_analysis(method = 'wilcox.test',estimate_group = 'Group') |>
EMP_mutate(wilcox = pvalue,
wilcox_fdr = fdr,
mutate_by = 'feature',location ='rowdata') |>
EMP_diff_analysis(method = 'DESeq2',.formula = ~Group) |>
EMP_mutate(DESeq2 = pvalue,
DESeq2_fdr = fdr,
mutate_by = 'feature',location ='rowdata') |>
EMP_filter(feature_condition = wilcox < 0.05 & DESeq2 < 0.05)
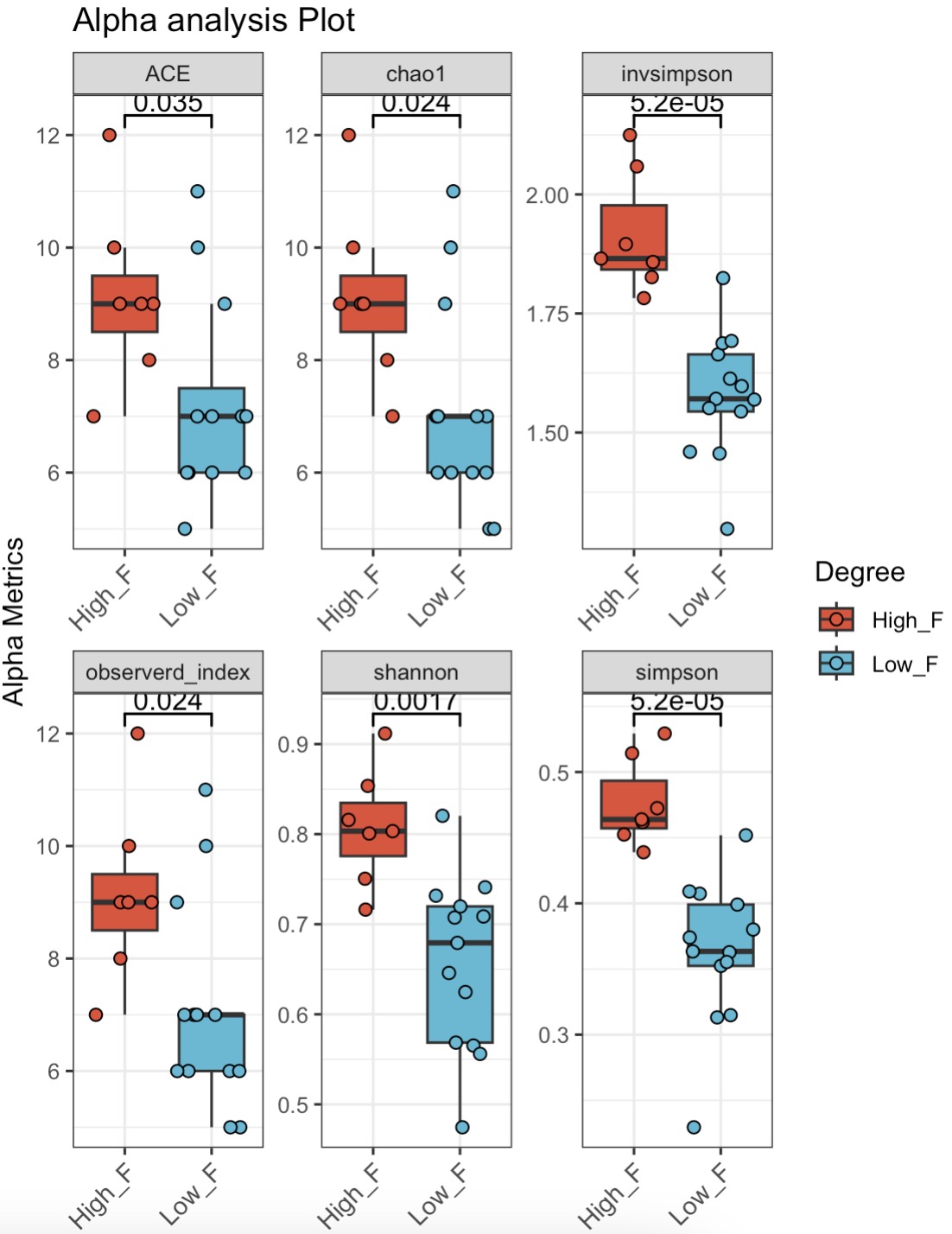
🏷️示例3:根据Firmicutes丰度高低分组,并进行alpha多样性比较
MAE |>
EMP_assay_extract('taxonomy') |>
EMP_decostand(method = 'relative') |>
EMP_collapse(collapse_by = 'row',estimate_group = 'Phylum') |>
EMP_mutate(Degree = dplyr::case_when(
Firmicutes >= mean(Firmicutes) ~ "High_F",
Firmicutes < mean(Firmicutes) ~ "Low_F"),
.after = Group) |>
EMP_alpha_analysis() |>
EMP_boxplot(estimate_group='Degree')